mm2-fast: Accelerating minimap2 for long-read
sequencing applications on Modern CPUs
Authors:
Saurabh
Kalikar, Intel Labs
Chirag Jain,
Indian Institute of Science, Bangalore
Vasimuddin
Md, Intel Labs
Sanchit
Misra, Intel Labs
Heng Li, Harvard Medical School
Highlights
1. Minimap2 is a widely used DNA sequence
alignment tool which supports many use-cases including mapping long reads or a
draft genome assembly to a reference sequence.
2.
Intel Labs, Indian Institute of
Science and Harvard Medical School have developed mm2-fast, an
accelerated version of minimap2, which optimizes end-to-end mapping time for
long read sequencing data by up to 1.8 times compared to minimap2 without any
loss of accuracy. This blog summarizes this work that is published at Nature
Computational Science https://www.nature.com/articles/s43588-022-00201-8.
3. mm2-fast accelerates the three main computational modules of minimap2:
seeding, chaining and pairwise sequence alignment
by applying several architecture specific optimizations, designing a
SIMD-based parallel chaining algorithm, and a learned index data structure.
DNA sequencing is the fundamental step in various genomics data
analytics pipelines. For example, DNA sequencing is being used in identifying
new variants and mutations of COVID-19 virus to study its pathogenicity and transmissibility. The first step in the process of
DNA sequencing is creating multiple copies of the DNA sequence to be read, and
then create small fragments of those copies by arbitrarily splicing them. In
the second step, DNA sequencers read these fragments, called reads, which are
stitched together to form the complete DNA sequence. Stitching of reads can be
done either by mapping them to a reference DNA sequence or by assembling them de
novo by utilizing the overlaps across reads.
Long-read
sequencing technologies from Pacific Biosciences (PacBio) and Oxford Nanopore Technology
(ONT) have made significant leaps in terms of read lengths and accuracy since
their introduction to the market. With their massive sequencing throughput (>
1 Tbp per day), they are ready to scale to large
population-scale studies. Software for processing long-read sequencing data also
needs to scale commensurately. Among the various steps involved in a
long-read-based variant calling pipeline, mapping long reads or assembly
contigs to a reference sequence is the most important and time-consuming step. More specifically, this step searches for best matches of DNA sequences
of lengths ranging from a few thousands to a few millions of letters in another
DNA sequence of length of the order of a few Billion letters. Minimap2 is by
far the most widely used tool for this task with more than 400,000 downloads
and is the standard.
mm2-fast:
our accelerated version of minimap2
mm2-fast
is a result of collaboration between Intel Labs, Indian Institute of Science
and Prof. Heng Li (Harvard Medical School), who is the author of minimap2. In
this work, we have identified and accelerated the three most time-consuming hot-spots in minimap2 on modern CPUs. These three key
computational modules in minimap2 - (i) seeding, (ii)
anchor chaining, and (iii) pairwise sequence alignment - account for 85% to 97%
of the total mapping time. Acceleration of the seeding stage is achieved by
replacing the standard hash-table lookup with a machine learning based lookup
using a hardware-efficient implementation of learned index data structure. Acceleration
of the anchor chaining step is achieved by designing a single-instruction multiple-data
(SIMD) based parallel chaining algorithm which uses vector processing units (VPUs)
available on modern CPUs. In the final sequence alignment stage, the runtime has
been reduced by converting 128-bit (SSE) SIMD instructions to 256-bit (AVX2)
and 512-bit (AVX512) SIMD instructions.
Figure
1 depicts the workflow of minimap2 and improvements
applied in mm2-fast at various stages of the workflow. We have also ensured
that the final output remains 100% identical to minimap2, which allows users to
easily switch to a faster version of minimap2 whenever faster computing
throughput is desired.
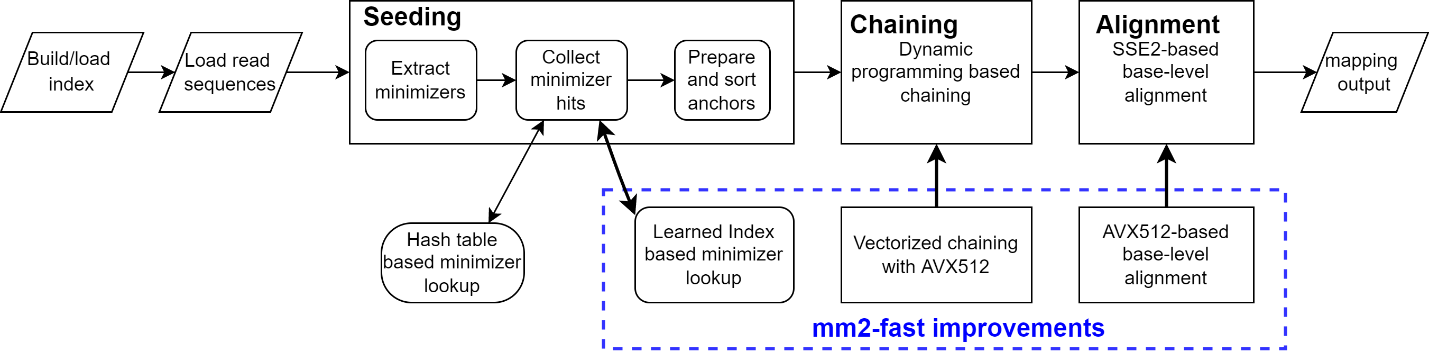
Figure
1: Minimap2 workflow depicting its three key modules: (i)
seeding, (ii) chaining, and (iii) alignment. Our improvements to each of the
modules are shown in the blue dotted rectangle.
To
showcase the wide applicability of our improvements, we benchmarked minimap2
and mm2-fast using three types of real human long-read sequencing data (ONT
Guppy 3.6.0, PacBio HiFi, PacBio CLR), and also three
human genome assemblies for mapping to the standard reference GRCh38. The
details of the datasets used are given in Table 1.
|
Query data set |
Genome sample |
Number of
reads/contigs |
N50 |
Maximum
length |
Source |
|
ONT |
HG002 |
19M |
50K |
543K |
|
|
HG003 |
24M |
44K |
760K |
||
|
HG004 |
29M |
48K |
1.1M |
||
|
PacBio Hifi |
HG002 |
8M |
13K |
30K |
|
|
HG003 |
7M |
15K |
32K |
||
|
HG004 |
7M |
15K |
31K |
||
|
PacBio CLR |
HG002 |
30M |
11K |
89K |
|
|
HG003 |
15M |
11K |
26M |
||
|
HG004 |
13M |
10K |
5M |
||
|
Genome Assembly |
CHM13 |
24 |
154M |
248M |
NCBI (GCA 009914755.3) |
|
HG002 (hap1) |
523 |
46M |
107M |
https://zenodo.org/record/4393631/files/ |
|
|
HG002 (hap2) |
507 |
40M |
131M |
https://zenodo.org/record/4393631/files/ |
Table1:
Description of datasets which were used to evaluate
mm2-fast. Each of these were mapped to GRCh38 human genome reference.
Figure 2 shows the
performance comparison of minimap2 (v2.22) and mm2-fast on a single socket Intel® Xeon® Platinum 8280
(Cascade Lake) Processor. mm2-fast outperforms minimap2 across all datasets and
achieves up to 1.76x speedup while keeping the output 100% identical to
minimap2. In other words, mm2-fast achieves faster
speed and reduced cloud computing costs without compromising accuracy. To the best of our knowledge, no prior work has reported
better end-to-end speedup of minimap2 using either a CPU, GPU, or FPGA.
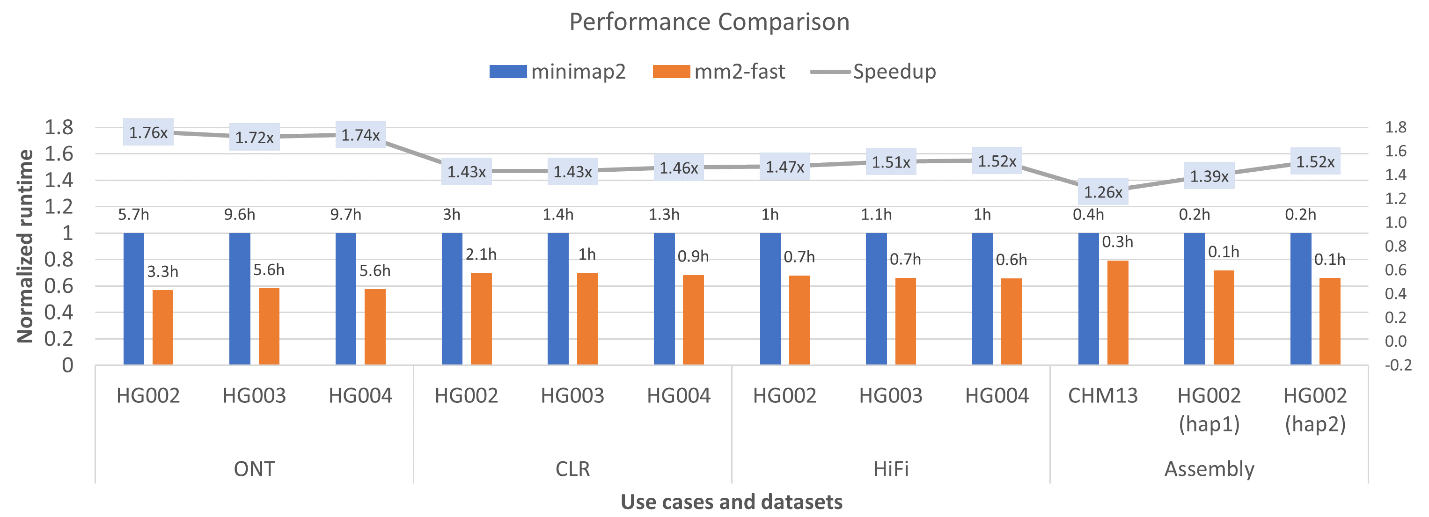
Figure 2: Performance
comparison of minimap2 and mm2-fast on a single socket Cascade Lake CPU (28
cores) for full datasets. X-axis shows various query datasets, y-axis is the
normalized time with respect to the mapping time taken by minimap2 corresponding
to each dataset. On top of the bars of minimap2 and mm2-fast, we show the
actual mapping time in hours by minimap2 and mm2-fast respectively. On the
secondary Y-axis, the line-graph shows the speedup achieved by mm2-fast.
Conclusions
The recent
advancements in long-read-based variant calling pipelines have shown a
promising future for the wide adoption of long read sequencing. As mapping long
reads to the reference sequence is the most important and time-consuming step,
we hope that mm2-fast will be seen as a faster alternative and drop-in
replacement of minimap2 for mapping long reads and this
work will benefit the wide scientific community engaged in long-read sequencing
projects.
mm2-fast is
published in Nature Computational Science (https://www.nature.com/articles/s43588-022-00201-8) and
the code is open-sourced on GitHub at https://github.com/bwa-mem2/mm2-fast. The current
version of mm2-fast is compatible with minimap2 v2.22 and mm2-fast will be
under active development as minimap2 further develops.
Configuration Details
Cascade Lake: 1-socket, 1x Intel(R)
Xeon(R) Platinum 8280 CPU, 28
cores, HT On, Turbo On, Total Memory 98 GB, 0x5000029, CentOS 22.04, Linux
release 7.6.1810 (Core), g++ (GCC) 9.2.0
Notices & Disclaimers
Performance varies by use,
configuration, and other factors. Learn more on the Performance
Index site.
Performance results are based on testing as of dates shown in
configurations and may not reflect all publicly available updates.
See backup for configuration details. No product or component can be absolutely secure.
Your costs and results may vary.
Intel technologies may require enabled hardware, software, or
service activation.
©
Intel Corporation. Intel, the Intel logo, and other Intel marks are
trademarks of Intel Corporation or its subsidiaries. Other names and
brands may be claimed as the property of others.